Toggle Dropdown
О проекте решения Совета Евразийской экономической комиссии «О внесении изменений в Правила проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза»
Распоряжение Коллегии Евразийской экономической комиссии от 9 июня 2020 года № 68 (г. Москва)
1. Одобрить проект решения Совета Евразийской экономической комиссии «О внесении изменений в Правила проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза» (прилагается) и представить его для рассмотрения Советом Евразийской экономической комиссии.
2. Настоящее распоряжение вступает в силу с даты его опубликования на официальном сайте Евразийского экономического союза.
Председатель Коллегии Евразийской экономической комиссии М. Мясникович
О внесении изменений в Правила проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза
Решение Совета Евразийской экономической комиссии от «__» _______ 20__ года №__
В соответствии с пунктом 2 статьи 4 и статьей 6 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года и пунктом 86 приложения № 1 к Регламенту работы Евразийской экономической комиссии, утвержденному Решением Высшего Евразийского экономического совета от 23 декабря 2014 г. № 98, Совет Евразийской экономической комиссии решил:
1. Внести в Правила проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утвержденные Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. № 85, изменения согласно приложению.
2. Настоящее Решение вступает в силу по истечении 6 месяцев с даты его официального опубликования.
Члены Совета Евразийской экономической комиссии:
От Республики Армения М. Григорян
От Республики Беларусь И. Петришенко
От Республики Казахстан А. Смаилов
От Кыргызской Республики Э. Асрандиев
От Российской Федерации А. Оверчук
ПРИЛОЖЕНИЕ
к Решению Совета
Евразийской экономической комиссии
от года №
ИЗМЕНЕНИЯ,
вносимые в Правила проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза
1. Пункт 6 указанных Правил изложить в следующей редакции: «При подтверждении биоэквивалентности лекарственных препаратов, которые выпускаются в лекарственных формах с модифицированным высвобождением, трансдермальных лекарственных формах и ингаляционных лекарственных формах, а также лекарственных формах для местного применения и липосомальных лекарственных формах исследования следует проводить в соответствии с требованиями приложений № 9 и 10, а также с актами, входящими в право Евразийского экономического союза (далее – Союз) в сфере обращения лекарственных средств.».
2. Приложение № 8 к указанным Правилам изложить в следующей редакции:
«ПРИЛОЖЕНИЕ № 8
к Правилам
проведения исследований биоэквивалентности
лекарственных препаратов в рамках
Евразийского экономического союза
(в редакции Решения Совета
от № )
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
ФАРМАКОКИНЕТИЧЕСКИХ ПАРАМЕТРОВ
|
Ae (0–t)
|
–
|
общее содержание неизмененного действующего вещества в моче, собранной от момента приема лекарственного препарата до времени t
|
|
AUC (0–72 ч)
|
–
|
площадь под кривой «плазменная концентрация – время» с момента приема лекарственного препарата до 72 ч
|
|
AUC(0 – ∞)
|
–
|
площадь под кривой «плазменная концентрация – время» с момента приема лекарственного препарата до бесконечности
|
|
AUC (0–t)
|
–
|
площадь под кривой «плазменная концентрация – время» с момента приема лекарственного препарата до последней определяемой концентрации во временной точке t
|
|
AUC (0– τ )
|
–
|
равновесная площадь под кривой в интервале дозирования
|
|
AUC (0–τ) ss
|
–
|
площадь под кривой «плазменная концентрация – время» в течение интервала дозирования в равновесном состоянии
|
|
AUC(t – ∞)
|
–
|
остаточная (экстраполируемая) площадь под кривой, определяемая по формуле 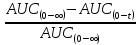
|
|
частичная AUC
|
–
|
частичная AUC, отделенная заранее выбранными точками отсечения
|
|
частичная AUC (х)
|
–
|
частичная AUC многофазных препаратов в фазу х
|
|
AUEC
|
–
|
площадь под кривой эффекта
|
|
C max
|
–
|
максимальная плазменная концентрация
|
|
C max,ss
|
–
|
равновесная максимальная плазменная
концентрация
|
|
C min , ss
|
–
|
минимальная плазменная концентрация в равновесном состоянии
|
|
С τ
|
–
|
концентрация в конце интервала дозирования
|
|
С τ, ss
|
–
|
концентрация в конце интервала дозирования в равновесном состоянии
|
|
D 1
|
–
|
длительность воздействия дозы, равная примерно 0,5 × ED 50
|
|
D 2
|
–
|
длительность воздействия дозы, равная примерно
2 × ED 50
|
|
Е 0
|
–
|
исходный эффект
|
|
Е max
|
–
|
максимальный эффект
|
|
ED 50
|
–
|
длительность воздействия дозы, при применении которой эффект составляет половину от максимального
|
|
k el
|
–
|
константа скорости терминальной элиминации
|
|
R max
|
–
|
максимальная скорость выведения с мочой
|
|
t 1/2
|
–
|
период полувыведения из плазмы
|
|
t max
|
–
|
время достижения C max
|
|
t max,ss
|
–
|
время достижения C max,ss
|
|
t lag
|
–
|
латентный период
|
|
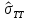 , , 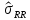
|
–
|
выборочная дисперсия
|
|
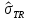
|
–
|
выборочная ковариация
|
|
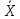
|
–
|
выборочное среднее».
|
3. Дополнить приложением № 9 следующего содержания:
«ПРИЛОЖЕНИЕ № 9
к Правилам
проведения исследований биоэквивалентности
лекарственных препаратов в рамках
Евразийского экономического союза
ТРЕБОВАНИЯ
к проведению фармакокинетического и клинического исследований биоэквивалентности кортикостероидных лекарственных препаратов для местного применения
1. Настоящие Требования содержат указания по подтверждению биоэквивалентности in vivo кортикостероидных лекарственных препаратов для местного применения путем проведения фармакодинамических исследований с использованием метода модифицированного биоанализа сужения сосудов по Стаутон-MакКензи (далее – биоанализ сужения сосудов, биоанализ побледнения кожи). Указанный метод предполагает оценку длительности воздействия для контроля дозы вводимых кортикостероидных препаратов для местного применения, а также проведение пилотного исследования зависимости «длительность воздействия дозы – ответ» для определения приемлемой длительности воздействия дозы в опорном клиническом исследовании, и проведение опорного клинического исследования биоэквивалентности in vivo с репликативным дизайном и подтверждением приемлемой зависимости «длительность воздействия дозы – ответ» субъектов. Как и все биоаналитические методики, данный фармакодинамический биоанализ требует детальной валидации, которая является обязанностью спонсора.
2. Сильнодействующие кортикостероидные лекарственные препараты для местного применения могут угнетать работу гипоталамо-гипофизарно-надпочечниковой оси, однако для препаратов, биоэквивалентность которых подтверждена в соответствии с настоящими Требованиями, представление результатов испытаний на подавление гипоталамо-гипофизарно-надпочечниковой оси в виде отчета в составе регистрационного досье лекарственного препарата не требуется.
3. Настоящие Требования применяются в отношении кортикостероидных лекарственных препаратов для местного применения независимо от уровня их активности. Поскольку характеристики зависимости «длительность воздействия дозы – ответ» могут изменяться в зависимости от конкретного лекарственного препарата, для определения соответствующих параметров основного (опорного) исследования рекомендуется проведение пилотного исследования.
4. Подтверждение биоэквивалентности 2 твердых лекарственных форм для приема внутрь обычно основывается на сравнении концентраций действующего вещества и (или) метаболита в доступной биологической жидкости (например, в крови или моче) после однократного или многократного дозирования каждого лекарственного препарата при проведении исследования с участием здоровых добровольцев. При невозможности применения этого метода для подтверждения биоэквивалентности разрешается использовать следующие методы исследований in vivo и in vitro (в порядке убывания предпочтительности):
а) фармакодинамические исследования;
б) клинические исследования;
в) исследования in vivo на животных;
г) исследования in vitro .
5. Для установления биоэквивалентности кортикостероидных лекарственных препаратов для местного применения в случае, если концентрация действующего вещества или его метаболитов не может быть оценена в доступных биологических жидкостях, требуется проведение фармакодинамического или клинического исследования in vivo . Клинические исследования обычно требуют включения большого числа субъектов и нередко не обладают достаточной чувствительностью. В отличие от них, фармакодинамические исследования позволяют получить приемлемые данные о биоэквивалентности при участии относительно небольшого количества субъектов.
6. Регистрация воспроизведенных кортикостероидных лекарственных препаратов для местного применения должна основываться в первую очередь на оценке фармакодинамических эффектов. Такой подход обусловлен свойством кортикостероидов вызывать побледнение кожи вследствие сужения микрососудов кожи. Данное свойство связано с количеством действующего вещества, поступившего в кожу, и может являться основанием для сравнения поступления действующего вещества из 2 потенциально эквивалентных составов кортикостероидных лекарственных препаратов для местного применения.
7. Несмотря на то, что существует несколько видов анализа сужения сосудов, общий метод основан на местном нанесении здоровым добровольцам кортикостероидного лекарственного препарата на период 6 – 16 часов, с последующей визуальной оценкой прошедшим подготовку ослепленным наблюдателем степени побледнения кожи по балльной шкале (0 – 3 или 0 – 4 баллов) в одной временно́й точке, как правило, через 2 часа после удаления препарата.
8. Настоящие Требования предполагают проведение 2 исследований in vivo (пилотного исследования зависимости «длительность воздействия дозы – ответ» и опорного клинического исследования биоэквивалентности in vivo ) для сравнения исследуемого и референтного лекарственных препаратов. Пилотное исследование характеризует зависимость «длительность воздействия дозы – ответ» в рамках модели определения Еmax и проводится исключительно с использованием референтного лекарственного препарата. Предпочтительный для подтверждения биоэквивалентности метод оценки длительности воздействия дозы основан на использовании 3 длительностей воздействия доз: ED50 , D1 и D2 . Сравнение исследуемого и референтного кортикостероидных лекарственных препаратов в рамках опорного исследования проводится на уровне, длительности воздействия дозы, приблизительно эквивалентном популяционной ED50 по результатам пилотного исследования. Чувствительность опорного исследования устанавливается посредством нанесения референтного калибровочного стандарта на 2 уровнях длительности воздействия дозы: D1 (калибровочный стандарт с более короткой длительностью воздействия дозы) и D2 (калибровочный стандарт с большей длительностью воздействия дозы). Следует устанавливать D1 равной примерно 0,5×ED50 , а D2 – 2×ED50 по результатам пилотного исследования. Каждый субъект выступает в роли «детектора» в таком исследовании, поэтому только данные тех субъектов, у которых отношение фармакодинамических ответов на дозе D2 к дозе D1 соответствует установленному минимальному значению, могут быть включены в анализ и подвергнуты статистической обработке для подтверждения биоэквивалентности in vivo .
9. Для целей настоящих Требований используются понятия, которые означают следующее:
«субъект, не ответивший на лечение» (nonresponder) – субъект, не проявляющий ответ на однократную длительность воздействия дозы референтного лекарственного препарата использованного в тех же условиях (с окклюзией или без окклюзии) в пилотном и опорном исследованиях;
«субъект ответивший на лечение» (responder) – субъект, проявляющий ответ на однократную длительность воздействия дозы референтного лекарственного препарата использованного в тех же условиях (с окклюзией или окклюзии) в пилотном и опорном исследованиях.
III. Исследование фармакодинамических эффектов:
10. При оценке результатов исследований эквивалентности уполномоченные органы (экспертные организации) государств – членов Евразийского экономического союза (далее – государства-члены) должны удостовериться в проведении исполнителем исследований при изучении эквивалентности методом анализа сужения сосудов кожи:
валидации и стандартизации метода анализа сужения сосудов кожи как биоанализа;
выбора в качестве наблюдателя – персонала, прошедшего подготовку с целью надлежащей оценки сужения сосудов.
1. Валидация и стандартизация метода анализа сужения сосудов кожи
11. Применение анализа сужения сосудов для оценки биоэквивалентности кортикостероидных лекарственных препаратов для местного применения основывается на предположении, что сосудосуживающие свойства кортикостероидов при местном применении могут быть использованы для разработки метода стандартного валидированного биоанализа. Результаты разработки и валидация биоанализа должны быть документально оформлены.
12. В процессе экспертизы регистрационного досье кортикостероидного лекарственного препарата необходимо проводить сопоставление результатов валидации ВЭЖХ или ГЖХ методики для количественного определения концентрации кортикостероидного лекарственного препарата в крови после введения определенной дозы с результатами валидации методики биоанализа сужения сосудов. При использовании биоанализа сужения сосудов ответ детектора ВЭЖХ или ГЖХ на известное количество действующего вещества заменяется наблюдаемым фармакодинамическим ответом (в данном случае сужением сосудов при использовании кортикостероидного лекарственного препарата для местного применения) на количество введенного действующего вещества.
13. В то время как в типичном анализе концентрации в крови или моче используется только один инструмент и «детектор», каждый субъект исследования с использованием метода фармакодинамического биоанализа выступает в роли «детектора», отвечающего на известное или неизвестное количество действующего вещества. Несмотря на фундаментальные отличия между стандартным анализом концентрации в крови или моче и биоанализом, многие принципы стандартизации и валидации сопоставимы.
14. Фармакодинамическая зависимость между дозой или концентрацией кортикостероидного лекарственного препарата и его исследуемым фармакодинамическим эффектом применима в биоанализе сужения сосудов при наличии оценки ее линейности. Несмотря на то, что существуют различные модели описания зависимости «доза – эффект», для биоанализа сужения сосудов применяется модель оценки Emax, или соответствующая сигмоидальная модель Emax, которая, рассчитывается по следующей формуле:












 Законодательство
Законодательство